
Органическая химия
Важные объявления
24.02.2022 Я не уверен, что в дивном новом мире, который строится на наших глазах, кому-то понадобится органическая химия. Но поскольку мы больше, к сожалению, совершенно ничего не умеем, то я всё же продолжу что-то здесь делать, просто чтобы без дела не сидеть. Может быть всё же кому-то понадобится в будущем, которое когда-нибудь обязательно наступит.
Приехали, конечная !!
Обновления на сайте
10.01.2023 – В качестве покаяния за эпический конфуз с задачей про енамин в последней контрольной я сделал страничку про енамины, довольно подробную, даже занудную, но, надеюсь, полезную.
23.12.2022 – Добавлена страница про ацетилен-алленовую перегруппировку, из которой кроме прочего можно узнать, что общего между пирожными типа эклер и ацетиленовым зиппером.
12.04.2022 – На страницу про переходные металлы добавлена вторая лекция с голосовым сопровождением.
19.03.2022 – Добавлен ответ про реакцию Лёйкарта-Валлаха и метод Эшвайлера-Кларка в Ответы
07. 03.2022 – Добавлена вкладка про реакцию Перкина на страницу методов в химии карбоновых кислот
03.2022 – Добавлена вкладка про реакцию Перкина на страницу методов в химии карбоновых кислот
01.01.2022 – С Новым годом! Эксперимент!! Добавлена страничка для того, чтобы можно было задать вопрос. Пока не знаю, что из этого выйдет, но попробуем. Ответы будут на страничке Ответы.
02.11.2021 – Нечто необычное
19.
Этот сайт создается для факультативной помощи студентам 3-го курса Химического факультета МГУ, поэтому его структура близко соответствует программе курса органической химии для основного потока, но содержание не повторяет эту программу, а дополняет ее, местами выходя за ее рамки.

Но – и это настоящий disclaimer: Сайт не является отражением официального подхода химического факультета МГУ и кафедры органической химии к изучению этой науки. Поэтому его нельзя цитировать и подавать как “так учат на химфаке МГУ”.
Этот disclaimer не означает, что автор сайта – какой-то отмороженный экстремист, которому противно всё официальное. Напротив, это обычная защита от экстремистов, которые, вычитав что-то, по их мнению, нетрадиционное, начнут писать про гнездо ереси в уважаемом университете. За содержание данного сайта несёт ответственность только автор сайта. На Химфаке МГУ работает много превосходных профессоров и преподавателей, и каждый из них может и должен учить так, как сочтет нужным. Не забывайте, что Университет – это не средняя школа и не военное училище: смысл образования в университете – не запоминание и тем более не зазубривание какой-то официальной доктрины, а помощь в формировании системы взглядов, вашей собственной системы взглядов, с которой вы уже и приметесь развивать науку дальше.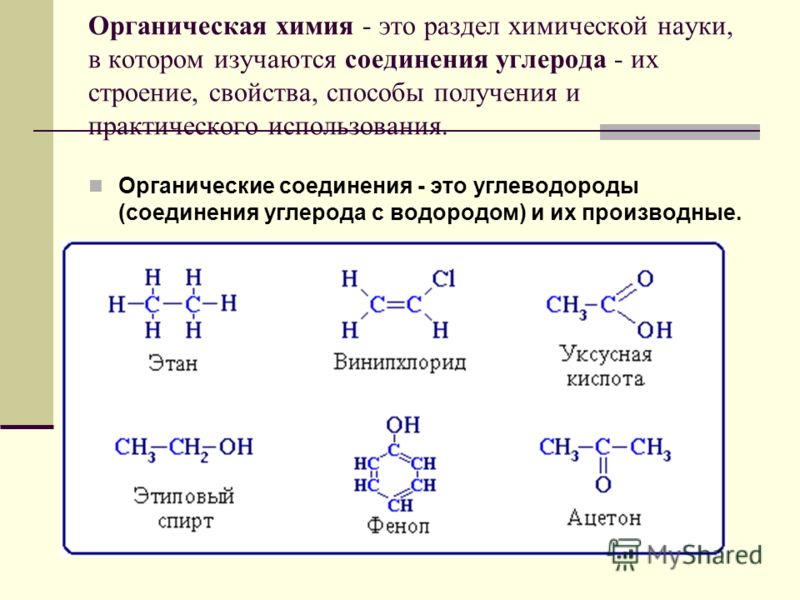

Механизм E2
Механизм Е2
Механизм E2, как и SN2, относится к согласованным механизмам. Напомню, что это означает, что исходные молекулы превращаются в продукты реакции напрямую, в 1 стадию, без участия каких-либо промежуточных частиц (интермедиатов). Суть механизма в том, что одни связи рвутся, другие одновременно образуются. Чтобы это произошло, все хозяйство должно перейти из одной ямы (минимума энергии соответствующего исходным) в другую (минимума, соотвесттвующего продуктам) через перевал, соотвествующий так называемому переходному состоянию. Энергия переходного состояния, всегда положительная, определяет энергию активации (если хорошо знаете хим кинетику, то не придирайтесь к терминам, здесь есть некоторые несущественные упрощения), а следовательно и скорость реакции. Чем ниже перевал, тем больше скорость, и наоборот. Изобразим все это:
Хорошо, механизм мы нарисовали, и что из этого следует? Так как механизм согласованный, то в самом важной стадии (а она тут одна) участвуют и субстрат, и основание. Поэтому здесь все важно: а) основность и размер основания; б) структура субстрата; в) качество уходящей группы. Самые важные следствия разобраны ниже (жмем на плюсики).
Поэтому здесь все важно: а) основность и размер основания; б) структура субстрата; в) качество уходящей группы. Самые важные следствия разобраны ниже (жмем на плюсики).
Конкуренция отщепления и замещения: основность
Извините, раздел в работе
Конкуренция замещения и отщепления: структура субстрата
Извините, раздел в работе
Как влияет растворитель
Мы это уже разобрали в Нуклеофильности и основности. Коротко напомню, что факторы, увеличивающие основность те же, что увеличивают нуклеофильность. Но по мере увеличения основности E2-отщепление выигрывает конкуренцию у замещения. Поэтому применение спецрастворителей типа ДМСО или межфазного переноса к анионам, которые уже в воде имеют основность, сравнимую с основностью гидроксид-иона, дает почти стопроцентное элиминирование.
На это можно возразить – а как это в воде основность выше основности гидрокcид-иода? Разве такое основание неизбежно не оторвет протон от воды – и вообще мы все когда-то изучали, что собственная кислотность/основность растворителя задает потолок основности в данном растворителе. Поэтому мы начинаем страшно нервничать, когда видим в условиях какой-нибудь реакции метилат или этилат в воде.
Поэтому мы начинаем страшно нервничать, когда видим в условиях какой-нибудь реакции метилат или этилат в воде.
Все это верно и пересмотру не подлежит. Будем нервничать дальше и не позволим! Тем не менее, в таблицах основности можно отлично увидеть pK оснований, явно более сильных чем гидроксид. Это не экспериментальные величины, а просто оценки. Есть много всяких полуколичественных закономерностей, которые позволяют делать такие оценки из косвенных данных. А уж качественную оценку мы можем сделать сами, обычным образом обсудив влияние заместителей на основность, то есть, например, представив метилат как метил-замещенный гидроксид – индуктивный донор увеличивает основность, и т.п. Основность в каком растворителе? На самом деле, в любом – речь идет о том, что основность метилата в некотором растворителе выше основности гидроксида в том же растворителе. Но не в разных. В каждом растворителе мы имеем некоторый ряд основности, порядок в котором основность увеличивается или уменьшается.
Поэтому мы имеем право говорить, что метилат в воде более сильное основание чем гидроксид-ион. А значит, если мы знаем, что гидроксид в ДМСО или в условиях межфазного переноса только элиминирует, значит и от алкоксидов в ДМСО (межфазный перенос к алкоксидам применить труднее, так как там часто присутствует водная фаза, но при желании замесить твердый алкоксид калия с краун-эфиром в бензоле можно) трудно ожидать что-то иное.
Во всем остальном влияние растворителя на элиминирование не настолько велико, чтобы это нужно было обсуждать. Безусловно, когда мы обсуждаем элиминирование по Гофману с образованием наименее замещенного олефина из всех возможных, основной тезис в том, что сильные пространственно затрудненные основания предпочитают отрывать протон из наименее замещенного положения и таким образом способствуют именно этому направлению. Растворитель не изменяет пространственных препятствий, но основность может увеличивать. Можем сделать вывод, что если трет-бутилат в трет-бутаноле отщепляет в основном по Гофману, то трет-бутилат в ДМСО даст еще более высокую пропорцию Гофман/Зайцев (100 к нулю не бывает никогда, и мы реально будем сравнивать что-то типа 80 к 20 и 90 к 10). Но это не так важно. Цифры нас запоминать никто не просит, а в решении задач мы предполагаем, что выбор направления практически полный.
Но это не так важно. Цифры нас запоминать никто не просит, а в решении задач мы предполагаем, что выбор направления практически полный.
Стереохимия в элиминировании E2: анти- и син-элиминирование
Механизм E2 требует совершенно конкретной стереохимии. Это легко понять, если еще раз посмотреть на механизм. В отличие от двух стадийных механизмов в механизме E2 все происходит между исходными и конечными веществами практически одновременно, и все вовлеченные в процесс атомы должны выстроиться в наиболее благоприятную конфигурацию.
Нарисуем переходное состояние, снабдив атомы орбитальками, вовлеченными в процесс. Видим, что π-орбитали двойной связи образуются из бывших гибридных орбиталей, обслуживавших связи C-H и C-X. Наилучшим образом все это действие будет развиваться тогда, когда эти связи в исходной молекуле находятся в одной плоскости – тогда по мере разрыва этих связей орбитали плавненько переходят в орбитали π-связи. А это в свою очередь возможно в двух случаях (очевидна аналогия с присоединением, чтоочень удобно, так как присоединение и отщепление можно рассматривать как одну и ту же реакцию в прямом и обратном направлении):
- син – это когда H и X находятся с одной стороны
- анти – когда с разных сторон;
Из этой картинки не ясно, какой из этих путей лучше.
А вот син-элиминирование требует фиксации в заслоненной конформации, что, для подавляющего большинства нециклических соединений практически невозможно, такой конформации не соотвествует ни один конформер, потому что такому расположению атомов соотвествует максимум энергии. Молекула в такой конформации не бывает никогда. Поэтому, син-элиминирование для нециклических соединений строго говоря вообще не может происходить. В реальности все не так строго – син-элиминирование все же происходит, но выходы соотвествующих продуктов почти всегда существенно меньше, чем выходы продуктов анти-элиминирования, настолько, что в планировании реакций син-элиминированием можно пренебречь.

Теперь перейдем к конкрентным случаям, с которыми придется справляться. Сразу обозначим важное ограничение – стереохимию отщепления анализируют только для E2-элиминирования по Зайцеву. В элиминировании по Гофману стереохимия почти всегда не имеет значения, а с совсем экзотическими случаями мы никогда не столкнемся. Как всегда жмем на заголовок, чтобы посмотреть, что внутри.
В элиминировании по Гофману стереохимия почти всегда не имеет значения, а с совсем экзотическими случаями мы никогда не столкнемся. Как всегда жмем на заголовок, чтобы посмотреть, что внутри.
Название Реакции
Портал органической химии
Реакции
Пожалуйста, используйте следующий URL, если вы хотите установить ссылку: https://www.organic-chemistry.org/namedreactions/.
Товар месяца
A B С Г Д Ф Г Ч И Дж К Л М Н О П В Р С Т У В З X Y Z
а
Конденсация ацетоуксусного эфира
Синтез ацетоуксусного эфира
Конденсация ацилоина
Реакция ольдер-эн. b
Baeyer-Villiger Oxidation
Baker-Venkataraman Перегруппировка
Реакция Бальца-Шимана
Реакция Бэмфорда-Стивенса
Декарбоксилирование Бартона
Реакция Бартона-МакКомби (Бартон Desoxygenation)
Baylis-Hillman Reaction
Beckmann Rearrangement
Benzilic Acid Rearrangement
Benzoin Condensation
Bergman Cyclization
Bestmann-Ohira Reagent
Biginelli Reaction
Birch Reduction
Bischler-Napieralski Reaction
Blaise Reaction
Реакция Блана
Синтез пиридина Больмана-Ратца
Борная кислота Реакция Манниха
Восстановление Буво-Блана
Перегруппировка Брука
Браун гидроборация
Реакция Бюхерера-Берга
Перекрестная связь Бухвальда-Хартвига Реакция
c
Соединение Кадио-Ходкевича
Окислительное восстановление Канниццаро
Восстановление CBS
Соединение Чана-Лама
Конденсация Клайзена
Восстановление по Кляйзену 00003
Click Chemistry
Collins Reagent
Элиминация COPE
COPE перестройка
Реакция CONIA-ene
COREY-BAKSHI-SHIBATA РЕЗЕПЕНЬ
COREY-CHAYKOVSKY Реакция
COREY-FUCHS Реакция
COREY-KAYKOVSKY
COREY-FUCHS Реакция
COREY-KAYKOVSKY
COREY-FUCHS Реакция
COREY-KAYKOVSKY
COREY-FUCHS.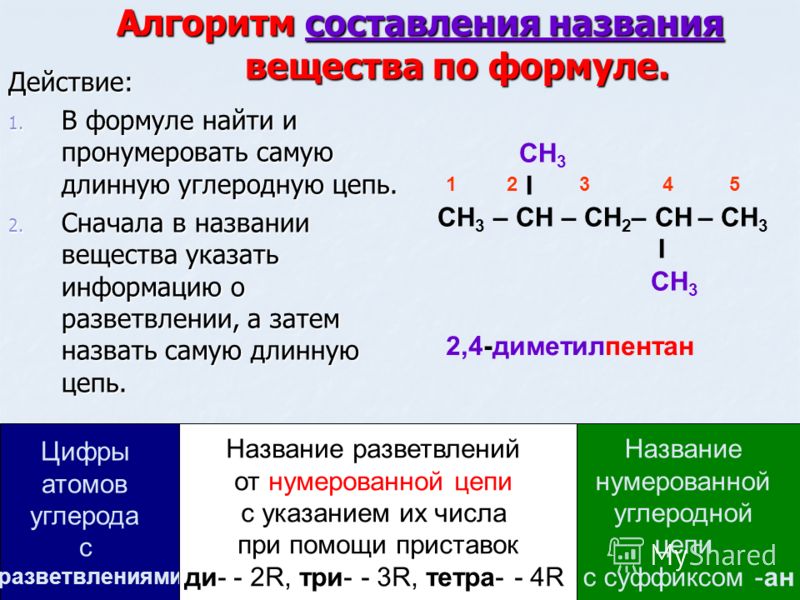 Реакция Кори-Зебаха
Реакция Кори-Зебаха
Реагент Кори-Саггса
Синтез олефинов Кори-Винтера
Синтез кумарина
Механизм Криги для озонолиза
Кросс-метатезис
Перегруппировка Курциуса (реакция)
D
DAKIN Реакция
DARZENS CONDEDSATION
DARZENS Реакция
Окивание Дэвиса
DE KIMPE Синтез азиридина
Delpine Реакция
DESS-Martin Oxitation
DIALISATION
DESS-MARTINATATION
DIALESTATION
DECKMANTATION DELMANSITATION
DECKMANTATION
DECKMATION
. DECKMANTATION
. ,3-Диполярное циклоприсоединение
Направленное орто-металлирование
Модификация Дёбнера
e
Реакция Эглинтона
Реакция Эне
Метатезис энина
Эпоксидирование
Реакция Эшвейлера-Кларка
Пиролиз сложных эфиров
Этерификация
f
Реакция Фаворского
Реакция Финкельштейна
Этерификация по Фишеру
Синтез индола по Фишеру
Окисление по Флемингу-Тамао
Ацилирование Фриделя-Крафтса
Алкилирование Фриделя-Крафтса
Синтез Фридлендера
Перегруппировка Фриса
Комбинация Фукуяма
Восстановление Фукуяма
2 gСинтез Габриэля
Реакция Гевальда
Соединение Глейзера
Коозонолиз Грисбаума
Реакция Гриньяра
Реакция Граббса
2
h
Галоформная реакция
Ганч Дигидропиридин Синтез (синтез пиридина)
Связка сена
Гек-реакция
Hell-Volhard-Zelinsky Реакция
Генри Реакция
Hiyama Coupling
Hiyama-Denmark Coupling
Hofmann Elemination 9000
HOFMANNNARNN. Реакция
Реакция
Реакция Хосоми-Сакураи
Циклоприсоединение Хьюсгена
Реакция Хунсдикера
Гидроборирование
i
Реакция Айрленда-Кляйзена
Восстановление Ицуно-Кори
Реакция Иванова (реагент)
j
Якобсен Эпоксидирование
Якобсен-Кацуки Эпоксидирование
Йоцик Реакция
Джонсон-Кори-Чайковский Реакция
Джонс Окисление
Джулия-Литго Олефинация
Юлия-Коциенски Олефинация
k
Реакция Кабачника-Филдса
Kindler Reaction
Knoevenagel Condensation
Kochi Reaction
Kolbe Electrolysis
Kolbe Nitrile Synthesis
Kolbe-Schmitt Reaction
Koser’s Reagent
Kowalski Ester Homologation
Kulinkovich Reaction
Kulinkovich-de Meijere Reaction
Kulinkovich -Реакция Шимоняка
Муфта Кумада
л
Реагент Лавессона
Лейкарт Тиофенол Реакция
Люче Редукция
m
Синтез малонового эфира
Реакция Манниха
Правило Марковникова
Реакция Макмерри
Меервейна-Понндорфа-Верлея Сокращение
Модификация Майерса Реакция Рамберга-Беклунда
Циклизация Майерса-Сайто
Аддишн Михаэля
Реакция Михаэлиса-Арбузова
Реакция Мицунобу
Реакция борилирования Мияуры
Модифицированная Юлия Олефинация
Добавление альдола Мукаямы
n
Циклизация Назарова
Реакция NEF
Негровая связь
Newman-Kwart перестройка
Реакция нитроальдола
NOZAKI-HIYAMA COUPLING
Ядерная подставка (S n
Ядрофильная подставка (S n
Ядрофильная подставка (S n
Ядерный. / S N 2)
/ S N 2)
o
O’Donnell Amino Acid Synthesis
Реагент Охира-Бестмана
Метатезис олефинов
Окисление Оппенауэра
Перегруппировка Овермана
Перегруппировка Окси-Коуп
Озонолиз
P
PAAL-KNORR FURAN Синтез
PAAL-KNORR PYRROLE SYNTTHESE
PAAL-KNORR THIOOPHEN SYNTTHES Олефинация
Реакция сочетания пинакола
Перегруппировка пинакола
Реакция Пиннера
Реакция Првоста
Реакция Прилежаева
Реакция Принса
1002Реакция Пшорра
д
r
Реакция Рамберга-Беклунда
Реакция Реформатского
Метатезис замыкания кольца
Метатезис раскрытия кольца (Полимеризация)
Реакция Риттера
Аннуляция Робинсона
Восстановление по Розенмунду
Реакция по Розенмунду-фон Брауну
Окисление по Руботому
S
Реакция Sakurai
Реакция Sandmeyer
Правило Saytzeff
Реакция Schiemann
Модификация Schlosser
Shmidt
Schotten-Baumann Реакция
Seebach Umpolung
Schotten-Baumann Реакция
Seebach Umpolung
Schotten-Baumann Реакция
Seebach umpolung
Schotten-Baumann Реакция
Seebach umpolung
Schotten-Baumann.
Sharpless Aminohydroxylation
Sharpless Dihydroxylation
Sharpless Epoxidation
Shi Epoxidation
Simmons-Smith Reaction
Sonogashira Coupling
Staudinger Cycloaddition
Staudinger Reaction
Staudinger Reduction
Staudinger Synthesis
Steglich Esterification
Stetter Reaction
Муфта Stille
Strecker Synthesis
Муфта Suzuki
Сверн Окисление
t
Окисление Тамао-Кумада
Олефинирование Теббе
Реакция Тищенко
Реакция Цудзи-Троста
Аллилирование Троста
u
Реакция Уги
Реакция Ульмана
Дигидроксилирование Апджона
v
Ван Лойзен Синтез имидазола
Ван Лойзен Синтез оксазола
Ван Лойзен Реакция
Викариозный нуклеофильный Замена
Реакция Вильсмайера
w
Wacker-Tsuji Oxidation
Weinreb Ketone Synthesis
Wenker Synthesis
Willgerodt-Kindler Reaction
Williamson Synthesis
Wittig-Horner Reaction
Wittig Reaction
[1,2]-Wittig Rearrangement
[ 2,3]-Перегруппировка Виттига
Реакция Воль-Циглера
Редукция Вольфа-Кишнера
Перегруппировка Вольфа
Вудворд цис – Гидроксилирование
Реакция Вудворда
Реакция Вюрца
Реакция Вюрца-Фиттига
х
y
Этерификация Ямагути
z
Почему существуют реакции на имена?
Реакции названы в честь первооткрывателей новаторских химических реакций или
уточнения ранее известных преобразований в том виде, в котором многие ученые
имеют свои имена, связанные с эффектом или явлением, уравнением, константой,
и т. д. В некоторых случаях человек, чье имя ассоциируется с реакцией, был
не первым, кто обнаружил реакцию, но вместо этого сумел ее популяризировать.
Имена реакций также могут просто описывать тип реакции, часто с использованием
инициалы или со ссылкой на структурные особенности.
д. В некоторых случаях человек, чье имя ассоциируется с реакцией, был
не первым, кто обнаружил реакцию, но вместо этого сумел ее популяризировать.
Имена реакций также могут просто описывать тип реакции, часто с использованием
инициалы или со ссылкой на структурные особенности.
Например, очень важной областью химического синтеза является углерод-углеродный
образование связи, и существует очень много именных реакций, описывающих такие
преобразования. В этой области разработка порядка использования
Магнийорганические соединения Виктора Гриньяра привели к совершенно новому дополнению
реакции, значительно расширившие возможности органического синтеза. В
исторический поворот, Гриньяр был не первым, кто использовал такие реагенты, а скорее
упростил процедуру, создав высокореактивный реагент на месте. Этот
популяризировал использование связанных преобразований, которые ранее
довольно утомительно, так как нужно было подготовить чувствительные магнийорганические реагенты
отдельно и хранится. То, что мы теперь знаем как реактивы Гриньяра, чаще всего используется
в дополнение к карбонильным соединениям, которые дают спирты или другие продукты в
высокие выходы, и этот процесс в настоящее время называют реакцией Гриньяра.
В другом важном случае многим реакциям образования связи С-С способствует
палладиевый катализ, что позволяет более эффективно использовать реагенты и многое другое
легкодоступные условия. Примером может служить синтез биарильных фрагментов,
субструктуры, которые часто встречаются в соединениях, представляющих интерес в медицине.
химия. Показателем полезности этих реакций являются названные
реакции для многих вариантов этих катализируемых палладием биарильных сочетаний
реакции стали общепринятыми уже через несколько лет после открытия, даже во времена
жизни соответствующих авторов, таких как (Макото) Кумада, (Джон Кеннет или
JK) Стилле и (Акира) Сузуки муфта. Другие химики, разрабатывающие конкретные
вариантов или улучшений или гибридных условий, их имена добавляются как
в муфте Кумада-Тамао-Корриу.
То, что мы теперь знаем как реактивы Гриньяра, чаще всего используется
в дополнение к карбонильным соединениям, которые дают спирты или другие продукты в
высокие выходы, и этот процесс в настоящее время называют реакцией Гриньяра.
В другом важном случае многим реакциям образования связи С-С способствует
палладиевый катализ, что позволяет более эффективно использовать реагенты и многое другое
легкодоступные условия. Примером может служить синтез биарильных фрагментов,
субструктуры, которые часто встречаются в соединениях, представляющих интерес в медицине.
химия. Показателем полезности этих реакций являются названные
реакции для многих вариантов этих катализируемых палладием биарильных сочетаний
реакции стали общепринятыми уже через несколько лет после открытия, даже во времена
жизни соответствующих авторов, таких как (Макото) Кумада, (Джон Кеннет или
JK) Стилле и (Акира) Сузуки муфта. Другие химики, разрабатывающие конкретные
вариантов или улучшений или гибридных условий, их имена добавляются как
в муфте Кумада-Тамао-Корриу.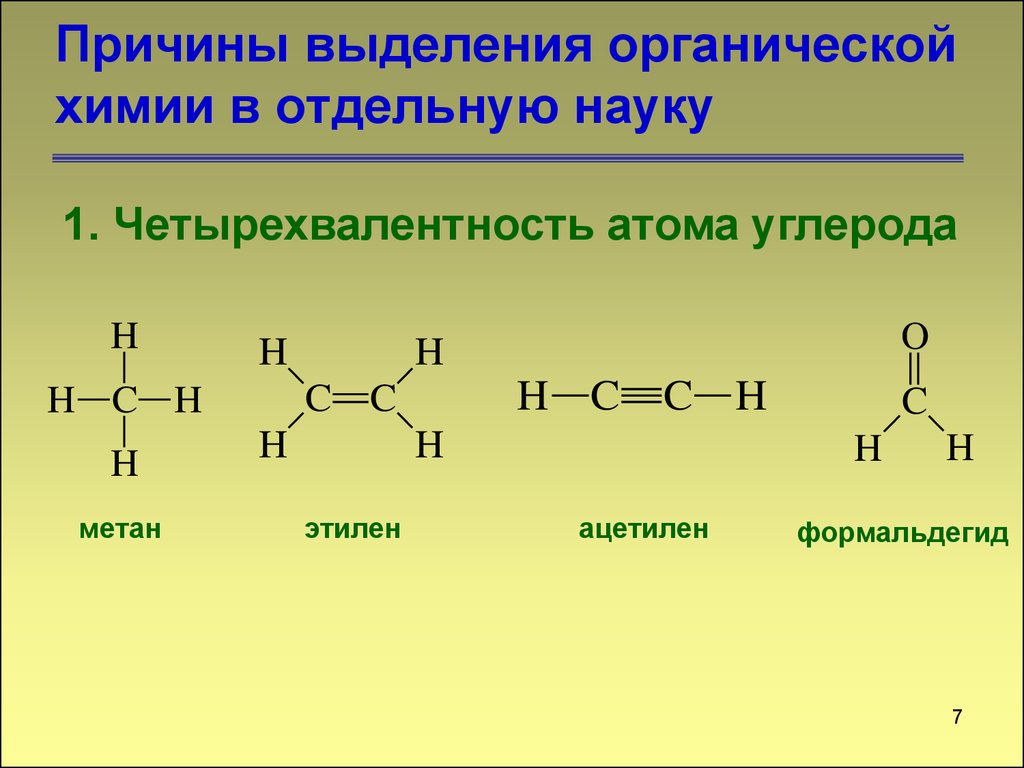
Помимо использования имен химиков, у нас также есть новаторские реакции, которые стали известны по аббревиатурам описательных названий, таких как «RCM» (метатезис с замыканием кольца) или INOC (внутримолекулярный нитрилоксид циклизация). Мы редко используем имя химика, разработавшего РКМ (Роберт Граббс) для обозначения реакции, но вместо этого признается его вклад применив его имя к используемым рутениевым катализаторам. Таким образом, мы говорим о «Граббсе». катализатор» или «катализатор Граббса 2-го поколения». Помимо таких названий, как «RCM», некоторые часто используемые реакции названы по особенностям строения предшественника или продукт. Примеры включают «альдольную реакцию» («альдол» — аббревиатура соединение, которое содержит как альдегидные, так и спиртовые функциональные группы) или «пинакол перестановка».
Почему мы должны учить десятки (или сотни!) реакций на имена?
Как упоминалось выше, реакции имен используются для обозначения новаторских
реакции или связанные с ними механизмы или принципы, которые стоит знать
и держать прямо. Точно так же, как врачи должны выучить названия органов и
геологи используют названия минералов, химики или студенты-химики используют названия
реакции как способ систематизировать свои знания и сообщить о химических
преобразования. В лабораторных дискуссиях люди очень часто используют реакции на имена.
ссылаться на эксперименты, которые они проводят, или на химические проблемы, которые они решают
расследование. Реакция на имя — это своего рода сокращение, позволяющее избежать необходимости
дать более подробное объяснение особенностей того или иного преобразования
интерес. Упоминание реакции на имя позволяет знающему слушателю донести
помнить о возможных субстратах, условиях реакции или деталях механизма.
Ожидается, что каждый в этой области знает наизусть базовый набор реакций на имена,
и это делает обсуждения менее трудоемкими. Таким образом, реакции на имена
стать частью общего словаря химиков органического синтеза. При встрече
коллега-химик, например, на конференции или во время собеседования, это
можно сделать первоначальную оценку уровня и глубины понимания вашего слушателя.
Точно так же, как врачи должны выучить названия органов и
геологи используют названия минералов, химики или студенты-химики используют названия
реакции как способ систематизировать свои знания и сообщить о химических
преобразования. В лабораторных дискуссиях люди очень часто используют реакции на имена.
ссылаться на эксперименты, которые они проводят, или на химические проблемы, которые они решают
расследование. Реакция на имя — это своего рода сокращение, позволяющее избежать необходимости
дать более подробное объяснение особенностей того или иного преобразования
интерес. Упоминание реакции на имя позволяет знающему слушателю донести
помнить о возможных субстратах, условиях реакции или деталях механизма.
Ожидается, что каждый в этой области знает наизусть базовый набор реакций на имена,
и это делает обсуждения менее трудоемкими. Таким образом, реакции на имена
стать частью общего словаря химиков органического синтеза. При встрече
коллега-химик, например, на конференции или во время собеседования, это
можно сделать первоначальную оценку уровня и глубины понимания вашего слушателя.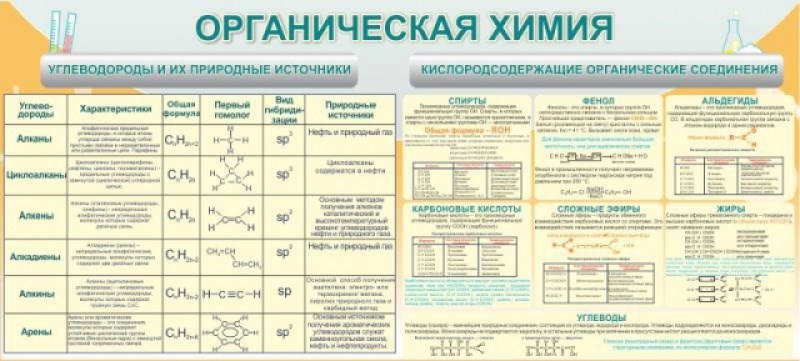 знания и опыт, ссылаясь на реакцию на экзотическое имя. Такой
признание может сигнализировать о том, что слушатель (или кандидат на работу) владеет
конкретной области химии. Это означает, что он или она будет способен
понимание деталей синтетических маршрутов в описанной работе и мог
возможно разработать альтернативы.
знания и опыт, ссылаясь на реакцию на экзотическое имя. Такой
признание может сигнализировать о том, что слушатель (или кандидат на работу) владеет
конкретной области химии. Это означает, что он или она будет способен
понимание деталей синтетических маршрутов в описанной работе и мог
возможно разработать альтернативы.
Органический синтез
Портал органической химии
Реакции > Поиск органического синтеза
Просмотр синтетических превращений по желаемому образованию связи
Графический указатель с различными опциями и ссылками должен помочь в разработка новых идей. Пожалуйста, попробуйте поиск по сайту напрямую, если вы не найдете желаемой реакции.
| Б | С 906:50 | Н | О | Си | Р | С | |
| Г / : | |||||||
| Б | |||||||
| С | |||||||
| Н | |||||||
| О | |||||||
| Si | |||||||
| Р | |||||||
| С | |||||||
| Ф | |||||||
| Класс | |||||||
| Бр | |||||||
| я | |||||||
| Сн | |||||||
| Хет | Гетероциклы | Гетероциклы | Гетероциклы |
Целевой синтез (TOS) и синтез, ориентированный на разнообразие (DOS)
TOS и DOS — это два совершенно разных подхода к органическому синтезу.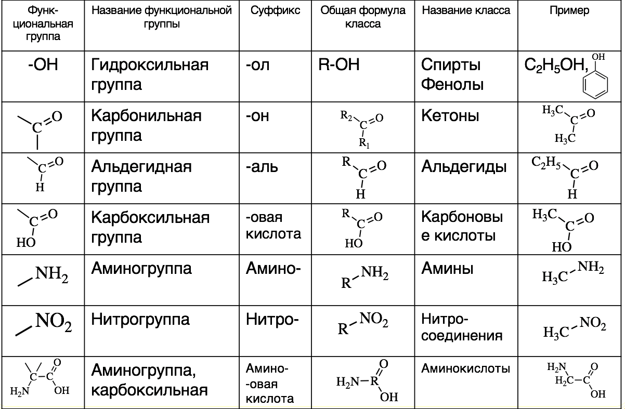 с разными целями и масштабами. Если конкретная целевая молекула, например, предлагает
интересующие свойства – такие как природное соединение с активностью против
конкретного заболевания – короткий, удобный и высокодоходный синтез должен быть
разработан, что позволяет быстро химически подойти к этой желаемой молекуле. В
В этом случае химик-синтетик берет лист бумаги или программу и пытается
разделить эту большую молекулу на более мелкие молекулы (разъединения) и повторить
эту процедуру итеративно, пока не появятся коммерчески доступные стандартные блоки.
Этот тщательный анализ, известный как ретросинтез, может привести к
несколько вариантов, которые приводят к нужной молекуле. В прямом синтезе
начиная с коммерчески доступных строительных блоков, несколько синтетических
затем необходимы шаги, пока желаемая молекула не может быть выделена. В
линейный подход, непредсказуемые узкие места могут привести к нежелательному возврату к
синтетическая отправная точка, поэтому доступно достаточно промежуточного продукта, тогда как
конвергентный подход ограничивает риск полного отказа, так как всего несколько шагов
синтез ветвей необходимо повторить.
с разными целями и масштабами. Если конкретная целевая молекула, например, предлагает
интересующие свойства – такие как природное соединение с активностью против
конкретного заболевания – короткий, удобный и высокодоходный синтез должен быть
разработан, что позволяет быстро химически подойти к этой желаемой молекуле. В
В этом случае химик-синтетик берет лист бумаги или программу и пытается
разделить эту большую молекулу на более мелкие молекулы (разъединения) и повторить
эту процедуру итеративно, пока не появятся коммерчески доступные стандартные блоки.
Этот тщательный анализ, известный как ретросинтез, может привести к
несколько вариантов, которые приводят к нужной молекуле. В прямом синтезе
начиная с коммерчески доступных строительных блоков, несколько синтетических
затем необходимы шаги, пока желаемая молекула не может быть выделена. В
линейный подход, непредсказуемые узкие места могут привести к нежелательному возврату к
синтетическая отправная точка, поэтому доступно достаточно промежуточного продукта, тогда как
конвергентный подход ограничивает риск полного отказа, так как всего несколько шагов
синтез ветвей необходимо повторить. В зависимости от таких рисков опытные химики-органики
запустить первые несколько реакций в большем масштабе и сохранить все промежуточные продукты,
поэтому они могут оптимизировать стадию реакции, если это необходимо.
В зависимости от таких рисков опытные химики-органики
запустить первые несколько реакций в большем масштабе и сохранить все промежуточные продукты,
поэтому они могут оптимизировать стадию реакции, если это необходимо.
Совершенно другой подход – это синтез, ориентированный на разнообразие. В ДОС,
химики-органики пытаются использовать разнообразные функциональные группы и позволяют
они реагируют с широким спектром субстратов, поэтому продукты охватывают широкий спектр
химическое пространство. Это включает в себя новые функции с совершенно другой привязкой
свойства, размеры колец и трехмерные конфигурации. Такие библиотеки, которые часто
созданный в комбинаторном подходе химии, может быть проверен против
множественные свойства (лекарственная активность, физико-химические свойства). Молекулы
которые показывают интересующие свойства (например, низкий IC 50 значение для
аффинность связывания с определенным ферментом) затем служат ведущими соединениями.